CUBI has developed many free and open source software packages. Below is a list of some highlights. For an exhaustive list, please refer to our GitHub repositories.
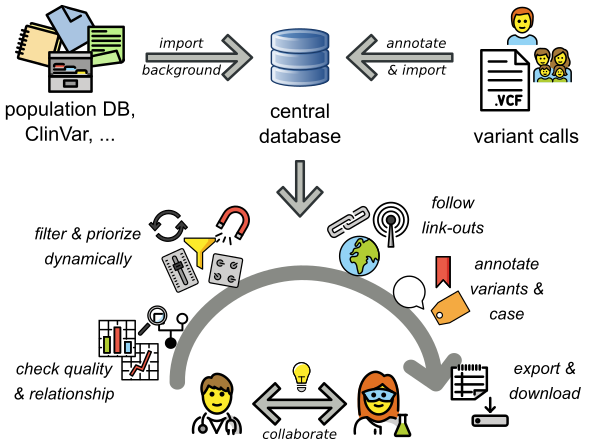
VarFish
VarFish is a user-friendly web application for the quality control, filtering, prioritization, analysis, and user-based annotation of DNA variant data with a focus on rare disease genetics.
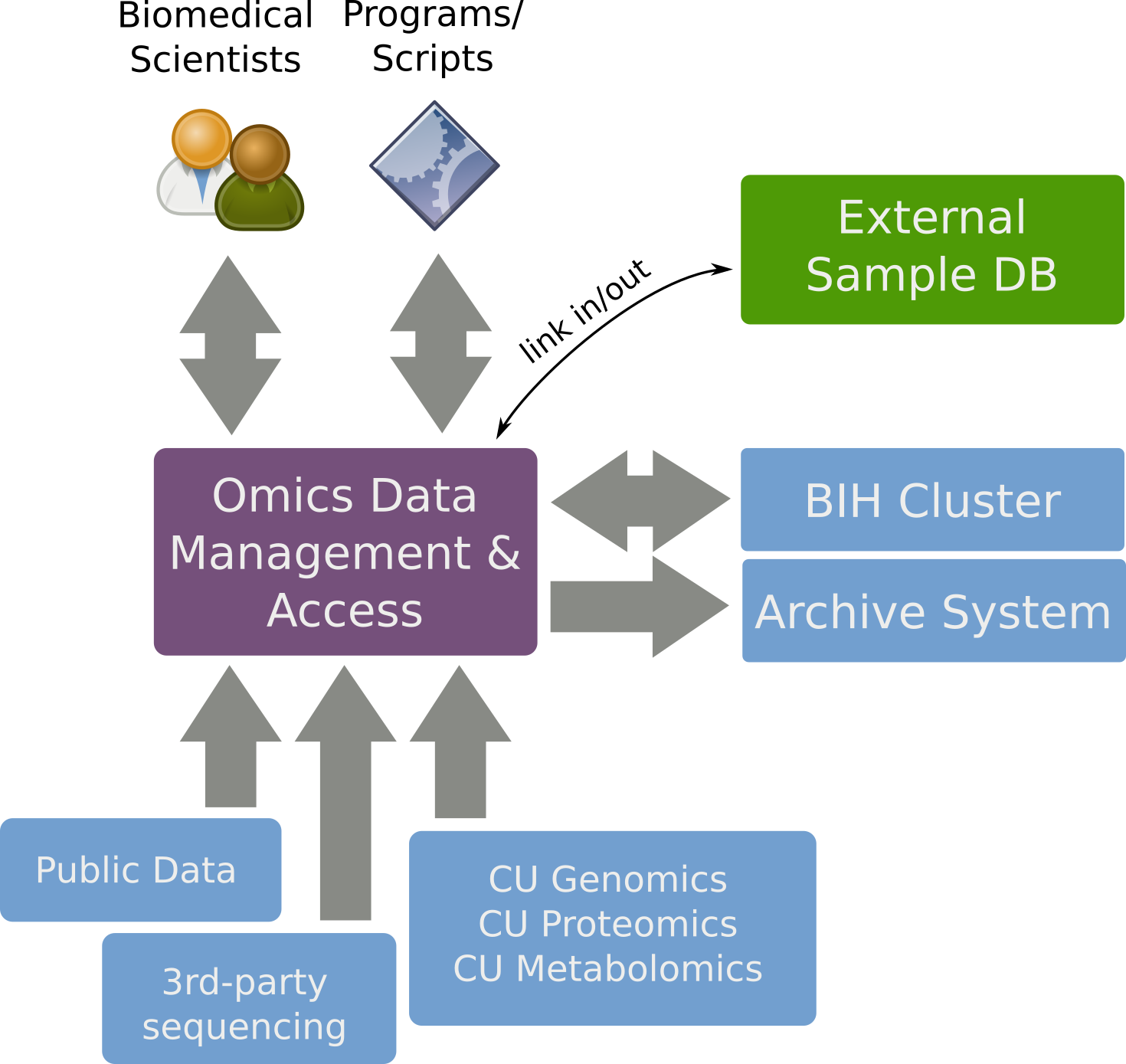
SODAR
SODAR provides
- FAIR data management for Omics studies,
- a web interface for biomedical scientists,
- command line interface and API for bioinformaticians.
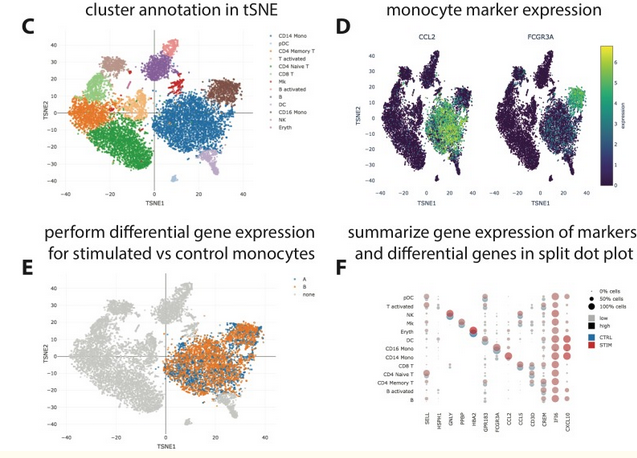
SCelViz
To facilitate analysis and interpretation of single-cell data by users without bioinformatics expertise, we present SCelVis, a flexible, interactive and user-friendly app for web-based visualization of pre-processed single-cell data.
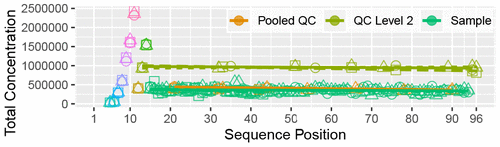
MeTaQuaC
Targeted quantitative mass spectrometry metabolite profiling is the workhorse of metabolomics research. Robust and reproducible data are essential for confidence in analytical results and are particularly important with large-scale studies.
NeatMS
Available automated methods for peak detection in untargeted metabolomics suffer from poor precision. We present NeatMS which uses machine learning to replace peak curation by human experts. We show how to integrate our open source module into different LC-MS analysis workflows and quantify its performance. NeatMS is designed to be suitable for large scale studies and improves the robustness of the final peak list.
DigestiFlow
Digestiflow is a software-package for the web-based management of Illumina flow cells.
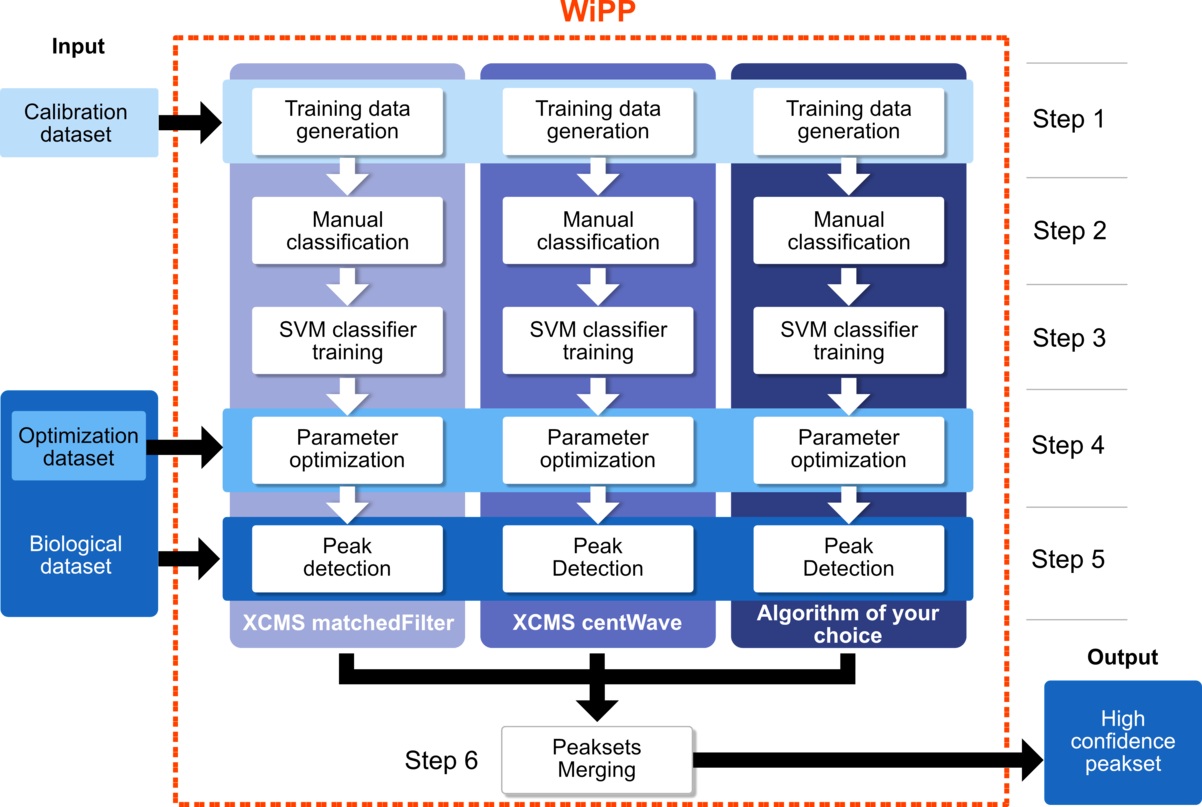
WIPP
Workflow for improved Peak Picking (WiPP) introduces a novel approach to automatic peak picking in GC-MS data in order to optimise the accuracy and quality of the process by combining the strengths of multiple existing or new peak picking algorithms.
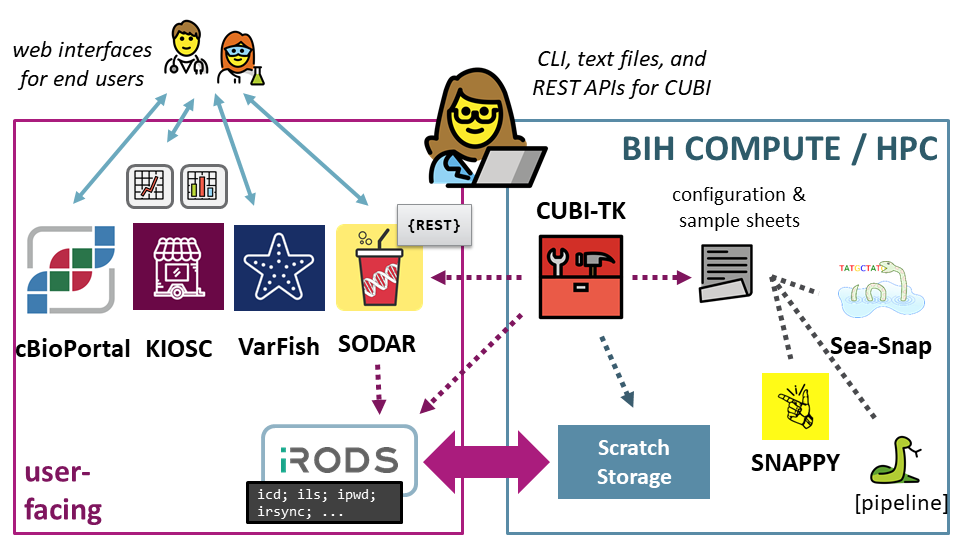
CUBI Toolkit
State-of-the art translational Bioinformatics, as practiced by CUBI, must employ a large and growing number of systems and services. Data must be importable from different sources, meta data must be captured and managed, data is processed with various specialized pipelines, stored in central systems, and delivered using different platforms. CUBI Toolkit is a command line application for the translational Bioinformatician at CUBI to glue all of these disparate parts together.
seaPiper
Standard processing of bulk RNA-Seq or pseudo-bulk scRNA-Seq data combined with gene set enrichment analysis produces a large amount of results. Static representations of such data are hard to navigate and do not provide the possibility to inspect and retrace the results. seaPiper is an interactive interface to present results of differential gene expression analysis, gene set enrichments and more.
tMOD
Gene set enrichment analysis is an important tool linking results of statistical analyses of high throughput data with biological reality. The basic idea is that if you group genes into gene sets (for example, interferon stimulated genes) you will be able to answer questions such as: are interferon stimulated genes up-regulated in COVID-19? The R package tmod is a collection of algorithms, tools and visualisation methods for gene set enrichments.
HLA-MA
Method HLA-MA for consistency checking in human HTS data analysis. Provided that there is sufficient coverage of the HLA loci, comparing HLA types allows for simple, fast and robust matching of samples from whole genome, exome and RNA-seq data.

VCFPy
VCFPy is a Python 3 library with good support for both reading and writing VCF.
clearCNV
A set of command line tools to call copy number variants on short read targeted sequencing panel data up to exome data. clearCNV can also re-assign samples to groups if the groups' data were generated on different panels but were confused or insufficiently documented.